easYPipe ‘prep’¶
Important
This step is a first mandatory step for the preparation of the data.
Usage¶
easypipe.py data prep [-h]
Example:
$ easypipe.py PROCESSED_DATA prep
How the data should be organized ?¶
The data folder (whatever it’s name) must contain only datasets folders.
Within each dataset folder, the processed data can be organized in several ways:
a mtz file directly in dataset folder
a mtz file in a sub-folder, or in a sub-sub-folder … of dataset folder
several processes are possible for a dataset, better if they are in different sub-folders, but not mandatory
if several mtz files are present in the same sub-folder, only the ones fitting the templates (from EDNA processes) will be treated, or if none fits only the first mtz file will be considered

Note
Data downloaded with easYGet are directly in the right tree organization.
What does it do ?¶
In an ‘easYPipe’ folder created at the place where it is executed, ‘prep’ copies each processed data mtz in a sub-folder of the dataset in this way:
creation of an ‘easYPipe’ treatment directory where it is run
creation of a subdirectory ‘0_processed_datasets’ where all the datasets folder are created
creation of a ‘data’ folder in each dataset folder and copy in this folder of processed mtz and log files
if there are several mtz in a folder, search for ‘EDNA’ treatment template and selects the right mtz files
Note
if you add a process for a dataset after a first ‘prep’, you can launch ‘prep’ sub-command again, this process will be added to the processes already copied
Then:
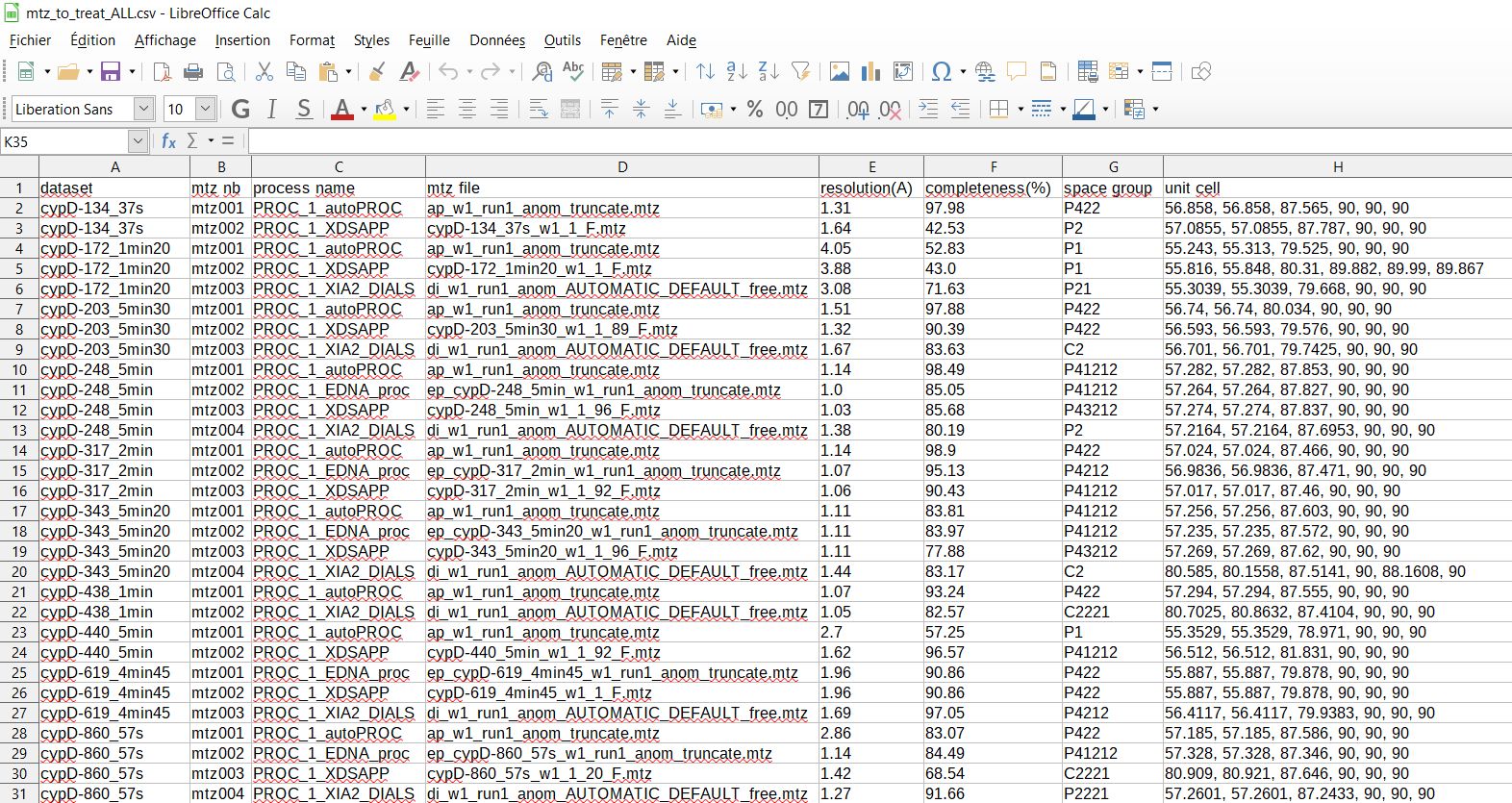
information on mtz files to be treated written in ‘/easypipe/1a_prep/mtz_to_treat_ALL.csv’ file
-creation of a csv file ‘/easypipe/1c_ligands/ligands_for_datasets.csv’ for future ligand generation with eLBOW [2]
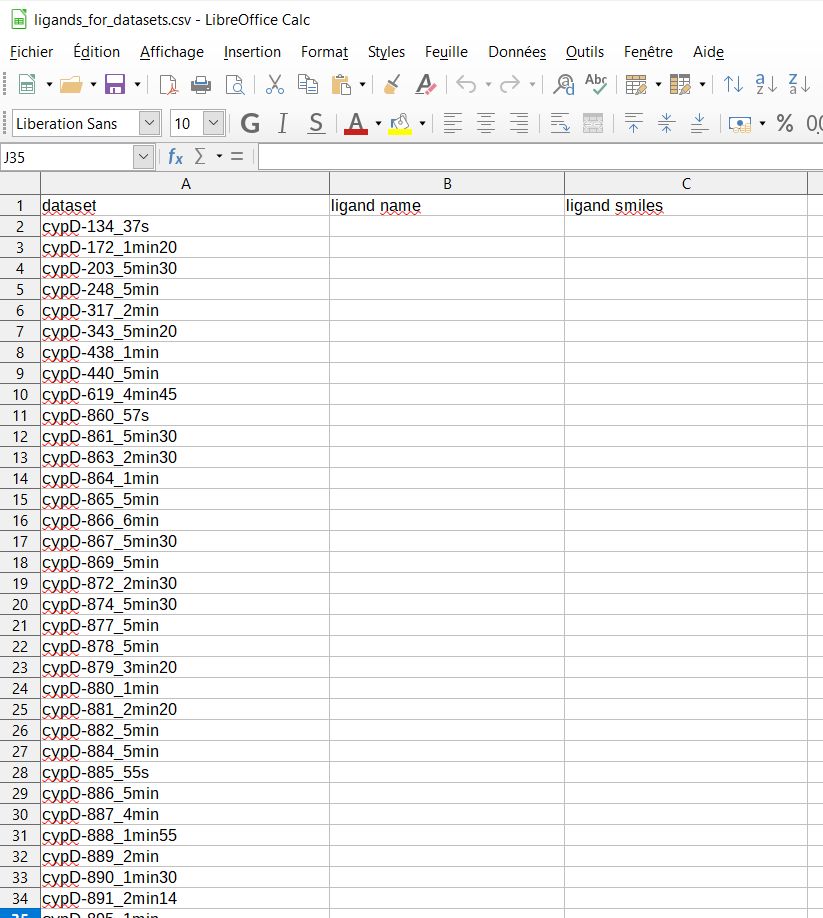
You have to fill ‘ligand name’ and ‘ligand smiles’ fields before running ‘easYPipe ligands subcommand’.
Caution
Save the modified csv file somewhere else or with another name if you don’t want to overwrite it in case you launch ‘prep’ sub-command again …
You can also run ‘easYPipe reindex subcommand’ if some mtz should be in higher symmetry space group.
If you are not interested in ligand placement or reindexation, you can directly run ‘easYPipe launch subcommand’.