easYPipe ‘launch’¶
‘launch’ subcommand runs phenix.ligand_pipeline [1] on all the mtz (several processed data, several datasets) according to options and information in ‘mtz_to_treat_ALL.csv’ file.
Usage¶
easypipe.py data launch [-h] [-m {fast,full,allsg}] [-l] [-n NUMBER] [-c NUMBER] [-b NUMBER | -a] [-s] [-t TEMPLATE] ref
arguments |
description |
|---|---|
ref |
folder with fasta file and pdb file for replacement, and cif(s) if ligand(s) in the model |
Warning
reference pdb files should include the row starting with ‘CRYST1’ containing information on space group
optional arguments |
description |
|---|---|
-h, –help |
show this help message and exit |
-m {fast,full,allsg}, –mode {fast,full,allsg} |
running mode: fast, full, or allsg (default = fast) |
-l, –lig |
for ligand search and placement |
-n NUMBER, –nblig NUMBER |
number of ligand copies to be searched (default = 1, max 9 for the moment). |
-c NUMBER, –cclig NUMBER |
minimum CC to consider a ligand placement correct (default = 0.7). Ligands with at least this CC will be incorporated into the current model for refinement. |
-b NUMBER, –best NUMBER |
launch only for mtz with best completeness, NUMBER indicates how many mtz to treat (default 1), ex: –best 2 |
-a, –autoproc |
launch only for mtz from autoPROC, or if none launch for mtz with best completeness |
-w, –whole |
launch for the whole mtz processes |
-s, –simulate |
only simulate, generate a csv file according to the future launch options. Give the possibility to modify the csv file to choose not to launch certain treatments, before launching again without simulation mode. |
-t TEMPLATE, –template TEMPLATE |
optional template name for log files and result folders, in case re-launching with different reference pdb of the same space group (else will be treated in existing folder and not launched again since it already exists). |
Example:
$ easypipe.py PROCESSED_DATA launch my_ref_folder --mode full --lig --best 2 --cclig 0.6
equivalent to:
$ easypipe.py PROCESSED_DATA launch my_ref_folder -m full -l -b 2 -c 0.6
What does it do ?¶
1. Sort mtz files according to space group in reference pdb, and decreasing completeness¶

If there are datasets without any mtz to treat according to space group, these datasets are listed in another csv file (“datasets_without_mtz_<sg_ref>.csv”).
2. List mtz files according to option ‘best’, ‘autoproc’ or ‘whole’¶
Option example: --best 1 (default)
List only mtz with best completeness for each dataset.

Option example: --best 2
List only 2 first mtz, when exist, with best completeness, for each dataset.

Option example: --autoproc
List only mtz from autoPROC, or if none list mtz with best completeness, for each dataset.

Option example: --whole
Whereas it is not recommended because it is time demanding, for problematic data it could be usefull to treat the whole mtz processed. You also can launch –whole option in simulate mode, and choose for processes to be treated or not, before launching again.

3. List mtz files with mode and ligand information for running Phenix¶
Note
Phenix options for the different modes are specified hereafter.
- For each dataset, write in a ‘launch csv’ file:
if ligand cif file is present for search when asked
mode that will be launched depending on mode asked, the presence (or not) of ligand cif file and data quality
information in case mode is different from mode asked
result folder name
Limits for poor data: There are minimum limits to process in ‘full’ or ‘allsg’ modes. These limits can be modified in config.py file (after what easypipe should be reinstalled).
minimum completeness (default = 70%)
minimum resolution (default = 3.75)
Poor data will be treated in ‘fast’ mode.
Option examples:
Option example: --mode fast (default)
Phenix uses a simple rigid-body refinement for model placement, which is faster and most of the time sufficient if the input model is already close enough to the target structure.

Option example: --mode full
Phenix will try rigid-body refinement first, then run Phaser if the R-free is too high (>0.4), it will run AutoBuild after initial refinement only if R-free is greater than the max_r_free cutoff = 0.3.

Option example: --mode allsg
In this mode, mtz will be treated regardless of the space group. Phenix will run Phaser, then run AutoBuild after initial refinement only if R-free is greater than the max_r_free cutoff = 0.3.

Option example: --mode full –lig
Phenix will be run in ‘full’ mode. Then ligand will be searched with LigandFit [2] and placed if cutoff model-to-map CC is more than 0.7 (default). This cutoff can be changed with ‘–cclig’ option. The number of ligands to be placed (default=1) can be changed with ‘–nblig’ option.

4. Launch Phenix according to chosen mode and options - Simulation mode¶
phenix.ligand_pipeline [1] is launched for each mtz file according to chosen mode and options, as listed in the ‘launch csv’ file (see 3. above).
If this ‘launch csv’ exists and you have modified something like adding a ligand cif for example, ‘launch’ mode should be run again, but in simulation mode so as it generates a new correct launch csv file instead of using existing one. When a new ‘launch csv’ file has been generated, just run the same command without simulation mode.
Example:
$ easypipe.py PROCESSED_DATA launch my_ref_folder --mode full --lig --autoproc --simulate
then:
$ easypipe.py PROCESSED_DATA launch my_ref_folder --mode full --lig --autoproc
Simulation mode also allows to modify the ‘to treat’ column of the ‘launch csv’ file (replacing ‘yes’ by ‘no’). Useful if you want to run some options only on some mtz. Then just run the same command without simulation mode. You can also modify the following columns: ‘mode’, ‘ligand search’, ‘CC’, ‘nb ligands’, as long as you know what you are doing.
5. Write results¶
At the end of each ‘launch’ subcommand, results are copied in a ‘RESULTS’ folder.
In datasets folders, copy of:
corresponding processed data and logs (useful for deposition at the PDB)
pdb and mtz result files
phenix cif file if ligand found
ligand folder, if exists
pdb of ligand(s) placed by LigandFit (all CC)

In a ‘ALL_pdb_mtz’ folder, for a rapid and easier visualization of all results in a same coot session, copy of all the pdb and mtz files of datasets treated in this ‘RESULTS’ folder.
In a ‘_mtz_treated’ folder, copy of:
csv listing datasets without mtz file
csv with mtz list
csv with mtz list after reindexing
csv with mtz list sorted according to reference space group
all ‘launch’ csv files, with a counter at the end of the names in case of several launches (with handmade modifications of launch csv file for example)

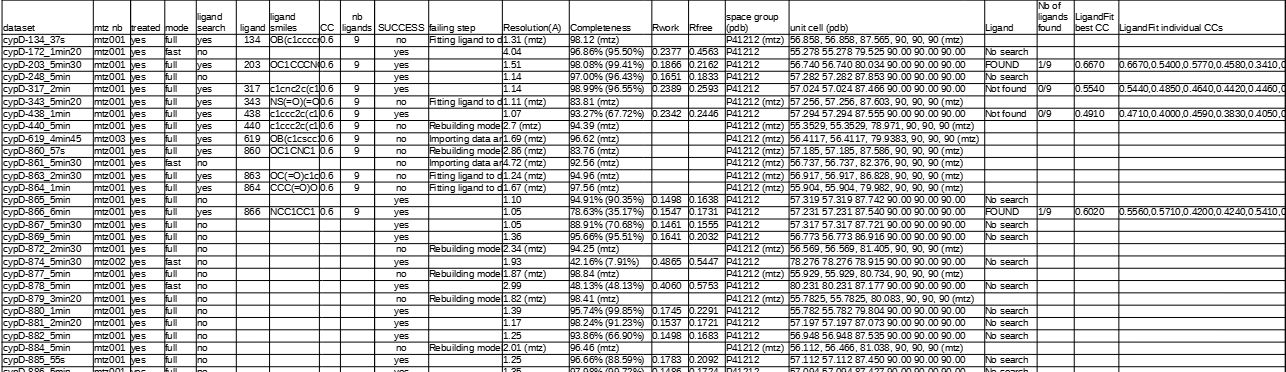
For each ‘launch’ subcommand, a csv file is created that summarizes the corresponding results for each dataset, with information on:
success of Phenix
failing step (in case success = no)
resolution (from pdb file, if failed from mtz data file)
completeness (from pdb file, if failed from mtz data file)
Rwork / Rfree
space group (from pdb file, if failed from mtz data file)
unit cell (from pdb file, if failed from mtz data file)
if ligand has been placed, number of ligands found, corresponding CC

Option example: -a –mode full –lig –nblig 9 –cclig 0.6
Finally, the results of all ‘launch’ subcommands you have run are compiled by running automatically the ‘summary’ subcommand.
Note
If there are several RESULTS folders (case when launched for several space groups, or different templates), a global SUMMARY file that compiles all SUMMARY files can be created by manually running ‘summary’ subcommand.
Phenix options according to modes (only for information)¶
phenix.ligand_pipeline [1] options are the following:
common options:
nproc=Auto
preserve_chain_id=True: Preserves the original chain ID
refine.after_ligand.hydrogens=False: Hydrogen atoms won’t be added prior to the final refinement step (else refinement significantly slower)
prune=False: disable Prune the model after refinement to remove residues and sidechains in poor density
keep_hetatms=True: prevent Phaser from resetting HETATMs occupancies to zero
refine.after_mr.update_waters=False: don’t add/remove waters automatically
‘fast’ mode:
skip_xtriage=True
mr=False: rigid-body refinement will be used
quick_refine=True: which will shorten both refinement steps from 6 to 3 cycles, and disable weight optimization.
build=False
skip_ligand=True
reference_structure=’model.pdb’: If specified, phenix.find_alt_orig_sym_mate will be applied to map the solution to the reference structure (not working when Phaser with several monomers)
‘full’ mode:
mr=Auto: the program will try rigid-body refinement first, then run Phaser if the R-free is too high (>0.4)
build=Auto: Run AutoBuild after initial refinement. By default, this will be done if R-free is greater than the max_r_free cutoff = 0.3
autobuild.quick=True: Run AutoBuild in quick mode. Inferior results, but a huge time-saver
quick_refine=True: which will shorten both refinement steps from 6 to 3 cycles, and disable weight optimization.
‘allsg’ mode:
mr=True
quick_refine=False
if ligand search:
ligand_copies=1 (except if option –nblig >1)
keep_input_restraints=True : if the input files include pre-calculated restraints for the target ligand, eLBOW will propagate these restraints instead of generating new ones.