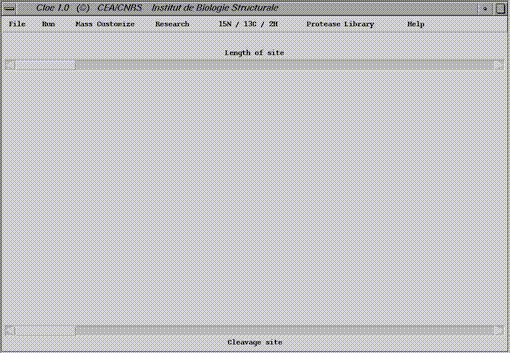
The open windows is called Main windows in the following section. If nothing is mentionned, the menu referes to the main window.
Cloe 1.0
Practical manual
P.Dosset, C.Vanbelle & D.Marion
Copyright 2004
Institut de Biologie Structurale J.P.
EBEL
CEA-CNRS
Laboratoire de Resonance Magnetique
nucleaire
Grenoble
The basic steps
Cloe can be launched either by double-clicking on the cloe.jar archive or by typing the command java -jar cloe.jar from the command line:
The open windows is called Main windows in the following section. If nothing is mentionned, the menu referes to the main window.
The basic steps.
Input File.Cloe can read a sequence directly from a text file ( Open file ) or the sequence can be created from scratch ( Define Sequence ).
Open file : File Menu -> Open File:Cloe handles only text files. Cloe reads only character correponding to an amino-acid in the one letter code. It is then not necessary to format file before use.
The file is opened via theFile menu and with the command Open File. The Open File dialog box pops up to choose your file. The text file appears in a new windows : Input file.
All lines before the sequence begining have to be erased. The end of the sequence can be marked by // sign (like in Swissprot and TrEMBL original format) or by the end of the file. If there is any text between the sequence end and the file end, one has to skip it or to mark the sequence end with //. Writing in the Input File windows is allowed, without effect on the source file. Any classical editing functions (copy, paste...) are enabled from (Edit menu) or to the Input File windows.
The used sequence has to be validated with Menu (Input File window) command Use. The Input File windows is then closed.
The sequence can be seen or modified afterward via File menu and Open -View/Define Sequence command.
A fragment mass calculation is possible in the Input File windows (Trick )
Define Sequence : Menu Open-View/Define Sequence:The sequence can be define from scratch via the File menu and Open-View /Define Sequence command. An Input file windows pops up. The sequence is writting in the large square area manually or with the standard editing function (copy, paste) accessible in the Edit menu (Input file window). The end of the sequence is the end of the file.
The used sequence has to be validated with Menu (Input File window) command Use. The Input File windows is then closed.
The sequence can be seen or modified afterward by the same way.
A fragment mass calculation is possible in the Input File windows ( Trick )
TrickThe mass of a sequence part can be calculated in the Input File windows. The desired sequence is underlined and the calculation mode (See Run Menu section) is selected in the Calculation Mode menu of the Input File windows. The results appears in the line under the menu bar. Warning : the calculated mass is for the fragment without addition of the water molecule mass.
Definition of the cleavage siteA cleavage site is defined by the lenght of the target sequence (Lenght site ), and the position of the cleaved bond (Cleavage site ). The kind of residues in each position has to be determined ( residue property ). The cleavage site of the most used proteases is already programmed ( Protease library )
From scratch.First of all the length of the site and secondly the position of the cleavage are defined with the respective scrolling bars. The cleaved bond is marked by an arrow.
The properties of the target sequence are determined for each position.
The residue can be selected :
- namely with the top menu ( set to none by default).At least, the list of selected residues appear as activated button, if selected, or inactivated button, if not, for each position.
- as a familly of residues in respect to their physico-chimic caracteristics (hydrophobic or hydrophilic and aromatic or non aromatic) with the two others menues (set to indifferent by default).
- in regard ro their charge status by pushing the appropriate button (acidic, basic, or neutral).

An user cleavage site can be saved or loaded with the save or load profile command located in the Protease Library menu.
Protease libraryThe profile of the most popular proteases are pre-programmed, according to the PROWL database ( http://prowl.rockefeller.edu/recipes/contents.htm ). The summing-up for each is available in the PROWL file . Select one profile from the list of the Protease Library menu. The main windows is then updated.
The cleavage site defined, the calculation
mode is selected in the Run menu. The selection implies calculation starting.
Four modes are available : average
, monoisotopic
, 15N/13C/2H
, and custom
.
The amino-acid masses take in account
the natural abudance of the isotopes (
http://www.ionsource.com/Card/Mass/mass.htm
). The atomic masses used in this mode are given in the following table.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The molecular weight for the added
water molecule is set to 18.015 u.m.a.
The amino-acid masses are calculated
with the 100 % contribution for the following isotopes : 1H, 12C, 14N, 16O,
and 32S. The atomic masses used in this mode are given in the following
table.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The molecular weight for added water molecule is set to 18.015 u.m.a.
In this mode, the contribution of the following stable istopes (15N, 13C and 2H) is taking in account for the calculation of the amino-acids mass. For example, for an 98 % 15N labelling, the nitrogen contribution is setting to 0,98*15.0001(15N) + 0,02* 14.0030 (14N). The atomic mass of the others atoms (H, C, O, S) are those of natural abundance.
The labelling ratio can be combined for each stable isotpe.
The atomic masses used in this mode
are given in the following table.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The molecular weight for added water
molecule is set to 18.015 u.m.a, until the 2H % is set to 0. When another
value is assigned, the water molecular weight is assumed to be lineary dependant
of the 2H % . Its molecular weight is then calculated as 2*[% 2H *
2.0141 + (1-% 2H)*1.0078] + 15.9994.
Before running this calculation
mode,the isotopic ratio has to be set up via the
15N/13C/2H
menu.
Amino-acids mass type, for example all the W, can be modified by increment or decrement of mass, in reference to the average, monoisotopic or 15N/13C/2H mode. One can also modify an amino-acid mass by its position in the sequence, for example residue 25.
Before running this calculation
mode, the kind of custom has to be set up via the th
e Mass Customize
menu.
Only one calculation mode can be used by run. When calculation is finished, Result window pops up.
The protein sequence used for calcultation is recalled in this section. The residue number and the molecular formula is also provided here.
If the sequence is coming from a file, the name and path of the file is writing as an header.
The molecular weight of the full length protein is given for all calculation modes.
The parameters used for the 15N/13C/2H and custom modes are also notified.
If these options have not been set up, the molecular weight are the average
one.
The length of the site and the position of the cleaved bond are recalled in this section.
All the combinations tested by the algorythm are listed. The cleaved bond is figured by two stars.
The calculation mode applied is notified. The proteolysis results are listed in a 6 columns chart. The peptides are sorted by descending molecular weight order.
1st column : number of the peptide
2nd column
: its boundaries
3rd column :
amino-acid sequence
4th : molecular
weight of the peptide. The add of a water molecule due to the cleavage of
the peptidic bond is taken in account.
5th : length
of the fragment
6th : molecular
formula
The results can be saved as a text file or print via the Save and Print command of the Menu menu (Result window).
Writing in the file and use of the standard edition function are
enabled.
The isotope ratio is set up with Define command of the 15N/13C/2H menu. The dialog box contains 1 field for each stable isotope. Inside, the expecting labelling yield is entered. Note that 1.00 corresponds to 100%. After set up, close the window in the Menu menu.
Customized
mass means to increment or decrement the mass of residues in reference to
their value on a calculation mode. There are two customized ways : by
residues type
or by position. Both are accessible via the Mass command of the Mass customized
menu. A Mass window pops up.
Customize by residue type.If the mass of all W want to be increase or decrease by a certain amount, positive or negative value respectively. The Per Mass option (default one) has to be selected. Then enter the mass change in the W field. The calculation mode in which the change is applied has to be selected on the Modify Mode menu.
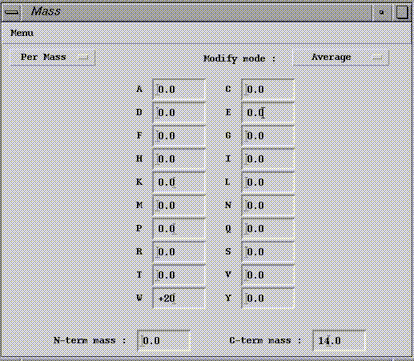
At the window bottom, there are two fields for the termini (N-term mass and C-term mass one). Enter there a mass modification to apply. In this case, the increment will be added to each proteolysis fragments.
If a residue position needs mass change, then select the Per Residue option. The dialog box contains 2 fields per line, one for position to modify (Num. residue) and the other for the mass change (mass). The calculation mode in which the change is applied has to be selected on the Modify Mode menu.
At the window bottom, there are two fields for the termini (N-term mass and C-term mass one). Enter there a mass modification to apply. In this case, the increment will be added to each proteolysis fragments.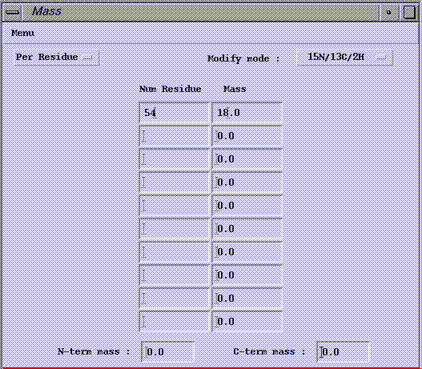
Two mass research modes are available : one to analyse proteolysis map MS data ( Research from mass ) and the other to calculate the mass of fragments from their limits ( research from boundary ).
To analyse MS data of
a proteolysis mixture, one has to take in account the cleavage protease site
and the experimental error. Cloe handles the both. The sequence is given
to Cloe (Input file
) and the cleavage site defined (definition of the
cleavage site
) . The option is opened with the From Mass command of the Research menu.
A From Mass windows pops up.
Note 1: because a digestion
can be incomplete, Cloe gives also fragments in which a target site is not
cleaved. This can be check in the amino-acid sequence displayed for each
fragment.
Note 2 : for the custom mode calculation, Cloe takes inaccount all paremeters, including the termini modifications.
Note 3 : t
he results can be saved as a text file or print via the Save and Print
command of the Menu menu (From mass window). Writing in the file and use
of the standard edition function are enabled.
For calculating mass wieght of fragment from their limits, activate the
From Boundary command in the reserach menu. A From Boundary window pops
up.

After filling the fields, select the desired calculation mode in the Run menu (of the From boundary window). The result appears in the result window. The struture of this one is exactly the same as previously described ( Result ) , except the proteolysis result replaces the research results.
The use of the program is subject to the following conditions:
1.The use of the program is restricted to the individual,
laboratory or organization to which it is
supplied. This individual,
laboratory or organization can make unlimited copies of the program
for backup purposes or
for running the program on more than one computer system at the host
institution.
2.Neither the program nor any part
of it may be sold, nor any copies distributed to third parties
without the express permission
of the authors at the Institut de Biologie Structurale.
3.Users may not decompile, disassemble,
reverse engineer or modify the program in any way.
4.No software package can be considered
to be bug free. The authors of the Cloe program
accept no responsability
whatsoever for damages resulting, directly or indirectly, from the
downloading and the use
of the program and make no warranty, either express or implied,
including but not limited
to, any implied warranty of fitness for particular purpose. The
Cloe program is
provided as it is and its users shall assume any loss, risk and damage when
using it.